Review Article, Issue 6.2
Sleep Apnea Phenotyping: Implications for Dental Sleep Medicine
http://dx.doi.org/10.15331/jdsm.7072
Victor Lai1, Jayne C. Carberry, PhD1,2, and Danny J. Eckert, PhD1,2
1Neuroscience Research Australia (NeuRA) and the University of New South Wales, Sydney, NSW, Australia; 2Adelaide Institute for Sleep Health (AISH), a Flinders University Centre of Research Excellence, Bedford Park, SA, Australia
ABSTRACT
New knowledge of obstructive sleep apnea (OSA) pathophysiology has highlighted the heterogeneity of this common chronic health condition. Recent advances in OSA ‘phenotyping’ concepts have provided a novel framework in which to understand OSA pathophysiology on an individual patient basis. This has also provided new potential precision medicine strategies to optimize efficacy and success rates with current OSA treatments including mandibular advancement therapy.
This review summarizes how different ‘phenotypes’ contribute to OSA pathophysiology and highlights the potential mechanisms by which mandibular advancement splints alter upper airway physiology according to an OSA phenotyping framework. In addition, it explains how understanding these phenotypes can facilitate novel and improved approaches to therapy, with a focus on phenotyping to improve mandibular advancement splint treatment prediction and efficacy. The potential to translate phenotyping concepts into the clinical setting is also discussed.
Keywords:
pathophysiology, precision medicine, sleep-disordered breathing, targeted therapy, upper airway
Citation:
Lai V, Carberry JC, Eckert DJ. Sleep Apnea Phenotyping: Implications for Dental Sleep Medicine. J Dent Sleep Med. 2019;6(2)
INTRODUCTION
Obstructive sleep apnea (OSA) is a chronic and prevalent sleep-related breathing disorder.1,2 It is characterized by recurrent, transient narrowing (hypopnea) and collapse (apnea) of the upper airway during sleep. These breathing disturbances cause frequent oxygen desaturations and sleep fragmentation. Up to 50% of men and 23% of women aged 40 to 85 years have moderate to severe OSA defined as 15 or more breathing disturbances per hour of sleep.1 The estimated prevalence of OSA has increased by 14% to 55% over the past two decades.2 Most individuals have OSA that is undiagnosed, untreated, or undertreated.3 This is a major concern given the extensive range of symptoms and adverse health consequences associated with untreated OSA. These include daytime sleepiness, decreased concentration, fatigue, irritability and memory loss,4 reduced quality of life,5 increased risk of motor vehicle accidents,6 cardiovascular disease,7,8 metabolic disorders,9 cognitive impairment,10 depression,11 and cancer.12 Thus, there is a pressing need for effective treatment to reduce the significant health and safety burden associated with untreated OSA.
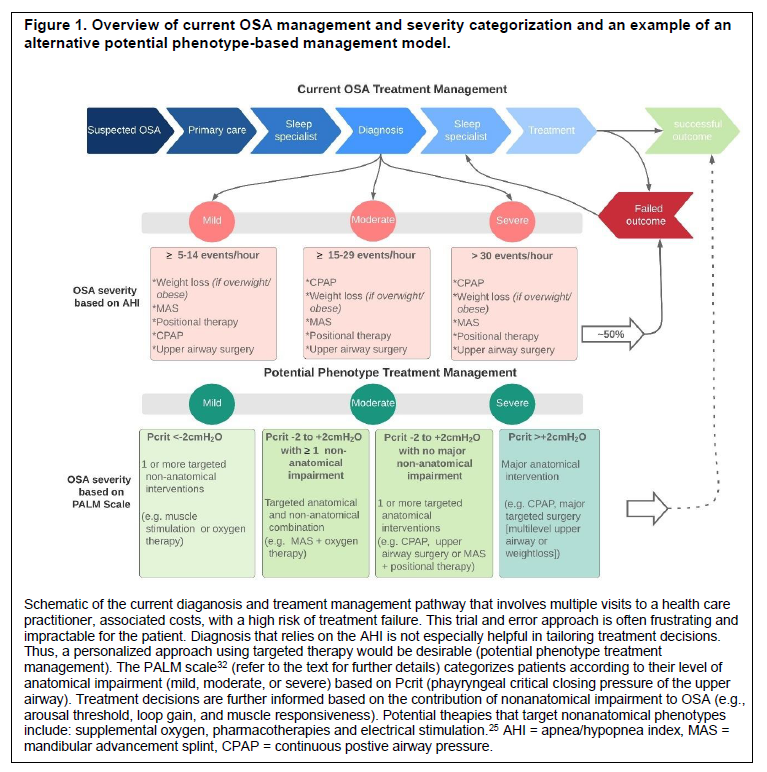
However, there are major challenges associated with the current management of OSA. Conventional therapies can be effective in reducing the severity and symptoms of OSA.13-16 Yet, fewer than 50% of patients adequately tolerate the first-line therapy, continuous positive airway pressure (CPAP).17,18 Oral appliances such as mandibular advancement splints (MAS) are a common alternative. However, efficacy is more variable than with CPAP and predicting which patients will respond to oral appliance therapy remains a major clinical challenge for the field.19,20 Indeed, various sophisticated assessments of upper airway anatomy during wakefulness and natural and drug-induced sleep as well as clinical, demographic, anthropometric and polysomnographic parameters have failed to consistently perform at satisfactory level to be incorporated clinically as prediction tools for MAS therapy outcome.19-23 Nonetheless, despite variable and unpredictable efficacy, oral appliance therapy can yield similar health benefits to CPAP.24 This is because lower efficacy compared to CPAP is typically counterbalanced by higher rates of adherence to therapy.24 One barrier to improved treatment efficacy with oral appliance therapy is that the precise mechanism or mechanisms of action are incompletely understood.19 This limits the ability to strategically alter oral appliance therapy design for improved efficacy and to target patients with clinical and physiological characteristics who are most likely to respond favorably. Other interventions for OSA are either difficult to achieve (such as weight loss) or also have variable efficacy (such as upper airway surgery or position therapy). Existing management guidelines rely on an inefficient trial-and-error approach that often begins with CPAP as the one-size-fits-all therapy.4,25 The current treatment journey is often costly, time consuming, and imprecise (Figure 1). As a result, many patients are lost to follow-up and are left untreated or undertreated.25 Thus, new treatment strategies that can accurately predict and optimize treatment outcomes for people with OSA are required.
Figure 1Overview of current OSA management and severity categorization and an example of an alternative potential phenotype-based management model. |
{kind=link}
The current diagnostic process for OSA also has limitations. The apnea-hypopnea index (AHI) is the main clinical measure used to define OSA severity. The AHI represents the number of breathing events per hour of sleep lasting ≥10 s that have either a ≥90% airflow reduction (apneas) from baseline or ≥30% airflow reduction (hypopnea) with an accompanying brief awakening from sleep (arousal) or oxygen desaturation (≥3%). Although a widely used metric, it has several shortcomings. The severity of OSA, as measured by total AHI, correlates poorly with the key symptoms and consequences of OSA.26-28 For example, a patient with longer but less frequent events may have a lower AHI than a patient with shorter but more frequent events.29 However, the patient with longer events may experience more severe hypoxemia and thus be more vulnerable to adverse cardiovascular events,30,31 despite having a lower AHI. Evidently, the AHI is inadequate to capture the heterogeneous presentations and consequences of OSA between individuals. Therefore, there is a need for new diagnostic approaches that can more comprehensively characterize OSA so that therapy can be tailored to the individual.
To address these diagnostic and treatment challenges, recent advances have been made in understanding the characterization and management of OSA through a ‘phenotypic’ approach.25,32,33 This precision medicine approach is the focus of the current review. More specifically, this review summarizes how different ‘phenotypes’ contribute to the pathophysiology of OSA and how understanding these phenotypes can unlock new avenues for targeted therapy. The role of phenotyping in MAS therapy to improve treatment and efficacy, and the potential to translate phenotyping concepts into the clinical setting is also covered.
A PHENOTYPIC APPROACH TO OSA PATHOGENESIS
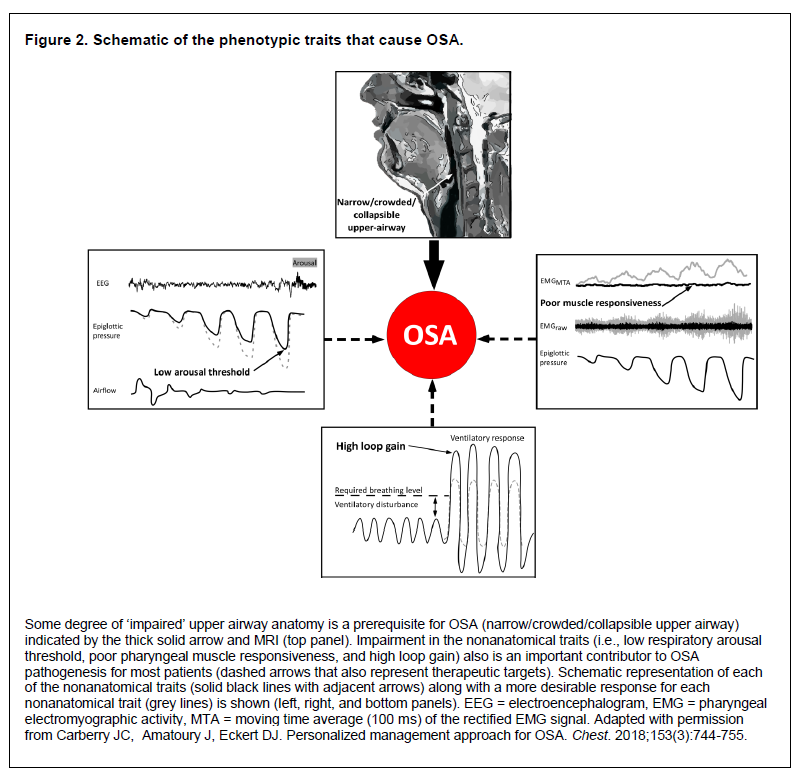
OSA is a multifactorial condition. Just as its clinical manifestations are heterogeneous, so too is OSA pathogenesis. There are multiple factors or ‘phenotypes’ that can combine in different ways to cause OSA (Figure 2). The heterogeneity of OSA pathogenesis is the reason that simple clinical measures, such as the AHI, are inadequate to characterize the multiple manifestations of OSA. This presents problems for current management approaches and the reliance on trial and error. However, the heterogeneity of OSA also presents an opportunity. Indeed, a more personalized approach has the potential to deliver targeted therapy or a combination of therapies to the individual as each phenotype represents a unique therapeutic target.25
Figure 2Schematic of the phenotypic traits that cause OSA. |
{kind=link}
Although it is likely that there are other important phenotypes or mediating mechanisms that have yet to be determined, to date, four major phenotypes that contribute to OSA pathogenesis have been identified. A narrow, crowded, or collapsible upper airway is the key phenotype.32,34,35 Indeed, some degree of underlying anatomical susceptibility to airway narrowing or collapse during sleep is essential for the development of OSA. However, the extent of impairment varies widely between patients, even in those with similar severity of OSA as measured by AHI.32 Moreover, the anatomical structures surrounding the airway remain constant between wakefulness and sleep and yet OSA only occurs during sleep. Thus, clearly there is more to OSA than just impairment in pharyngeal anatomy. Indeed, non-anatomical phenotypes are also key contributors to OSA pathogenesis. These include poor responsiveness or contractility of the upper airway dilator muscles during sleep; unstable respiratory control (high ‘loop gain’) causing large breathing oscillations; and a low respiratory arousal threshold to airway narrowing. Approximately 70% of individuals with OSA have one or more nonanatomical phenotypes that contribute to their OSA.32 The relative contribution of anatomical and each nonanatomical phenotype to OSA pathogenesis vary between individuals. This contributes to the heterogeneity of OSA, which can affect the clinical presentation and responses to therapy.
Other factors such as inflammatory processes, cerebral blood flow changes, hormonal changes, and postural alterations as well as currently unknown variables may also contribute to OSA pathophysiology and/or interact with these four OSA phenotypes.36 Nonetheless, the current phenotypic description of OSA pathogenesis, even if incomplete, has the potential to inform more tailored and comprehensive therapeutic approaches than current treatment management pathways.
THE KEY PHENOTYPE FOR OSA: ANATOMICAL IMPAIRMENT OF THE UPPER AIRWAY
In addition to the mandible, maxilla, and hyoid bones, which are rigid structures, the upper airway is composed of soft tissues such as the tongue, upper airway muscles, and parapharyngeal fat pads. This mix of bony support and soft tissues allow the upper airway to quickly change its shape and size to perform its various key functions, including swallowing and speech. However, the malleable quality of the upper airway also renders it vulnerable to closure and collapse during sleep in susceptible individuals.
Some degree of anatomical impairment in one or more of the components of the upper airway is necessary for the development of OSA. Imaging studies show that the cross-sectional area of the upper airway is reduced in individuals with OSA compared with those without OSA.34 This narrowing renders the upper airway susceptible to collapse, which can occur laterally, anteroposteriorly, or concentrically,37 at one or multiple levels along the pharynx.38 Nonetheless, the site just behind the soft palate (velopharyngeal area) is a particularly common site of collapse for most people with OSA.39 Obesity is a major contributor to a narrow upper airway. Fat deposition in the structures surrounding the airway such as in the tongue, soft tissues, lateral pharyngeal walls, and other pharyngeal muscles can reduce the upper airway space in individuals with obesity and OSA.40 However, up to half of all patients in whom OSA is diagnosed do not have obesity.41 Thus, anatomical factors such as retrognathia and smaller mandible area can restrict the size of the bony compartment of the upper airway to cause upper airway narrowing and closure during sleep independent of obesity for many individuals with OSA.42 Relatively smaller increases in weight in individuals of Chinese ethnicity compared to Caucasians also tend to favor increased propensity to OSA.43 This is caused, in part, by differences in craniofacial structures.43 Thus, in addition to obesity-related narrowing, the shape and position of the surrounding craniofacial structures can also contribute to a narrow or collapsible upper airway.34,44
Anatomical impairment affects the collapsibility or functional anatomy of the upper airway during sleep, measured using the gold standard passive critical closing pressure (Pcrit) technique.32 On average, individuals with OSA have a more collapsible or anatomically impaired upper airway than individuals without OSA (they have a higher Pcrit).32 Thus, their upper airway closes at higher pressures and requires increased levels of CPAP to stay open during sleep. However, the degree of anatomical impairment varies greatly among individuals with OSA with Pcrit values ranging from -5 cmH2O to more than +5 cmH2O.32,35 Indeed, within the subatmospheric range (Pcrit from -5 cmH2O to 0 cmH2O), there is considerable overlap in Pcrit values between individuals with and without OSA.32 For individuals with OSA with a moderate or mild anatomical impairment, there are additional nonanatomical phenotypes that contribute to OSA pathogenesis and mediate OSA severity. Nevertheless, anatomical impairment is still a key contributor to OSA and thus remains an important target for therapy. Indeed, most existing treatments – such as CPAP, MAS, weight loss, surgery, and positional therapy – focus on rectifying the anatomical impairment. However, as described earlier, in individuals with OSA these methods have limitations and provide suboptimal treatment or no treatment25 (Figure 1).
NONANATOMICAL PHENOTYPES AND THEIR ROLE IN OSA PATHOGENESIS
Poor Upper Airway Muscle Function
Because the pharynx lacks a rigid skeletal framework, upper airway patency is instead primarily maintained by the surrounding dilator muscles. These muscles receive complex neural drive from brainstem neurons that mediate different patterns of activation during quiet breathing.45 For example, the genioglossus, which is the largest upper airway dilator muscle at the base of the tongue, has increased activation during inspiration.45 The genioglossus also receives reflex input from pressure-sensitive mechanoreceptors in the upper airway as well as chemoreceptor input.46,47 Sleep onset48,49 and different sleep stages50 also alter the neural drive to dilator muscles and contribute to upper airway collapsibility during periods of low drive. However, there are other mediators of dilator muscle activity beyond sleep-dependent neural control that also contribute to OSA pathogenesis.
Even during sleep, respiratory stimuli (such as negative pharyngeal pressure changes or blood gas changes) can trigger increases in neural drive to increase upper airway dilator muscle activity.33,51 This response is referred to as muscle responsiveness. However, more than 30% of individuals with OSA have negligible muscle responsiveness to airway narrowing during sleep (experimentally induced via negative pharyngeal pressures changes).32 These individuals do not have adequate dilator muscle recruitment until very high levels of respiratory stimuli are reached. On the contrary, enhanced muscle responsiveness is protective against OSA, even in individuals with obesity who have anatomical impairment of the upper airway.52 Poor muscle effectiveness can also contribute to upper airway collapsibility in some individuals with OSA.33 This is the inability of dilator muscles to adequately dilate the upper airway in response to airway narrowing despite adequate neural drive and muscle activation. Causes include poor coordination of neural drive to dilator muscles (which can result in counterproductive movements),53,54 mechanically inefficient orientation of muscle fibers,54 or snoring-induced changes in the distribution of muscle fiber types (which can leave the upper airway more susceptible to fatigue).55,56 Thus, poor muscle responsiveness or effectiveness in individuals with pharyngeal anatomical impairment are important contributors to OSA pathogenesis (at least 30% of those with OSA).32 However, each component of the poor muscle function phenotype is potentially a novel target for therapy.
For example, upper airway muscle training can help reduce OSA severity. A systematic review showed that muscle training reduces the AHI by almost 50% and improves oxygen saturation and daytime sleepiness.57 However, efficacy varies widely among individuals. Thus, increased knowledge into the mechanisms of how pharyngeal training improves upper airway function is required to move beyond the current trial-and-error approach and to design targeted training regimes to optimize efficacy. Stimulation of the hypoglossal nerve, which provides drive to the muscles of the tongue, reduces the AHI by up to 70% or more with accompanying improvements in OSA symptoms.58,59 However, this approach is not suitable for all patients with OSA and up to one-third of those selected are nonresponders.58 As with any surgical procedure, potential safety and cost limitations should be considered. Pharmacotherapies to increase upper airway muscle activity have long been a therapeutic target for OSA. Recent studies have highlighted the potential importance of noradrenergic and anticholinergic targets.60 For example, the antidepressant desipramine prevents sleep-related reductions in genioglossus muscle activity and reduces upper airway collapsibility in healthy individuals and those with OSA.61,62 The combination of reboxetine and hyoscine butylbromide also improves upper airway function during sleep, albeit via different mechanisms than via increased genioglossus muscle activity.63 Most recently, combination therapy with atomoxetine and oxybutynin, which have strong noradrenergic and anticholinergic effects, increased genioglossus muscle responsiveness during sleep and reduced the AHI by approximately 75% (with all patients with OSA having a ≥50% reduction in AHI).64 Although further clinical trials are needed, pharmacotherapies are revealing novel and exciting potential approaches for management of OSA. This approach may prove to be especially fruitful in those with a poor upper airway muscle responsiveness phenotype.
Low Respiratory Arousal Threshold
It was previously thought that arousal from sleep was necessary to reopen the upper airway during a respiratory event.65 On the contrary, it is now understood that up to 75% of respiratory events in OSA are resolved without an arousal, or the arousal occurs after the respiratory event is resolved.66 In other words, arousals are not necessarily crucial for airway reopening and may be detrimental. Thirty percent to 50% of individuals with OSA wake from sleep to very small increases in breathing effort (as measured via negative esophageal or epiglottic pressure swings).32 These individuals are repetitively aroused from sleep to minimal stimuli and exhibit the phenotype of a low respiratory arousal threshold. This can contribute to their OSA pathogenesis for several reasons. One reason is that frequent arousals prevent deeper, more stable stages of slow wave sleep and destabilize breathing patterns as a result of rapid changes in blood gases, both of which can hinder adequate recruitment of upper airway dilator muscles.50,67 Indeed, as the stimuli for arousals are the same as those for upper airway muscle recruitment (negative pharyngeal pressure changes or blood gas changes), premature arousals limit the buildup of stimuli that is required to activate these muscles and reopen the airway.68 Accordingly, a low respiratory arousal threshold is another nonanatomical phenotype that can play a crucial role in OSA pathogenesis for some patients.Frequent arousals also lead to fragmented sleep, which can cause sleep deprivation and a common consequence of OSA, excessive daytime sleepiness. Common hypnotic drugs such as eszoplicone, zoplicone, zolpidem, and trazodone all raise the respiratory arousal threshold69-72 and can reduce the AHI by up to 50%69,70,73 in certain patients. Although genioglossus activity is not impaired,71,72 some hypnotic drugs at high doses may lengthen apneas and worsen oxygen saturation.71,74 In contrast, a recent 30-day randomized trial with zopiclone in patients with OSA with low and moderate respiratory arousal thresholds did not worsen overnight oxygenation or next-day sleepiness or objective alterness.75 However, there was also no therapeutic benefit on OSA severity.75 Thus, questions remain as to the risk versus harm profile of hypnotic drugs in OSA. This balance likely varies among patients according to their underlying phenotypes and between different classes of hypnotic drugs. Of the studies conducted to date, eszopiclone has yielded the greatest therapeutic benefit for OSA.70,76 However, a recent detailed physiology study with zolpidem showed improvements in two of the key phenotypic traits, an increase in the respiratory arousal threshold and also an unexpected threefold increase in genioglossus muscle responsiveness during sleep.72 Thus, this agent has therapeutic potential and additional studies are warranted.
High Loop Gain
A characteristic trait of OSA is the propensity to fluctuate between wake and sleep with periods of unstable breathing. Breathing during sleep is regulated by partial pressure of carbon dioxide, or PaCO2. A person’s sensitivity to changes in PaCO2 is mediated via negative feedback mechanisms that can be explained by the engineering concept of loop gain. In simple terms, loop gain is the ratio of the magnitude of a ventilatory response to a ventilatory disturbance. The key components that determine loop gain are: (1) plant gain (lungs, blood, and tissues where CO2 is stored in the body), (2) mixing gain (circulatory delay, i.e., the time it takes for a change in CO2 to mix with existing blood and be detected by the chemoreceptors), and (3) controller gain (sensitivity of the chemoreceptors, e.g. carotid body). A high loop gain (large ventilatory response to a small ventilatory disturbance) reflects an overly sensitive ventilatory control system. This phenotype perpetuates further breathing instability via excessive changes in CO2. Conversely, a more stable system occurs when the ventilatory response is proportionate to the disturbance.33,77 High loop gain is thought to play a key role in OSA pathogenesis for at least 30% of patients.32,78,79 Treatment strategies to lower loop gain and reduce the AHI (typically by approximately 50%) include supplemental oxygen and acetazolamide.80,81MAS THERAPY: MECHANISMS OF ACTION
To determine how MAS therapy can best fit into a personalized, phenotyping approach to treat OSA, the first crucial step is to understand exactly how MAS therapy alters the key OSA phenotypes. In addition to improved understanding of the mechanisms, phenotyping approaches should help to identify which patients with OSA will benefit most from MAS therapy.Upper Airway Anatomical Impairment
MAS devices protrude the mandible with the aim of stiffening the surrounding upper airway structures to prevent pharyngeal narrowing or collapse during sleep. Most studies to date have focused on how oral appliances alter upper airway anatomy and collapsibility (anatomical impairment). Imaging studies show that MAS therapy increases the total volume of the upper airway in responders to treatment.82-84 This is predominantly due to changes at the level of the velopharynx as reflected by increased cross-sectional area, especially in the lateral dimensions.82,83,85 A direct soft-tissue connection between the ramus of the mandible and the lateral walls of the velopharynx may account for these changes.86 Lateral displacement of the parapharyngeal fat pads may also contribute to the increase in cross-sectional area.82 Contrary to traditional thinking,87 MAS devices do not appear to systematically increase the oropharyngeal dimensions (section of upper airway at the level of the tongue).82 However, MAS devices can pull the entire tongue forward and prevent it from obstructing the upper airway in individuals with lower AHIs.86 MAS devices also elevate the hyoid bone.82 These anatomical changes are associated with improvements in OSA severity82 and reflect some of the key mechanisms by which MAS therapy improves upper airway anatomy. MAS devices also improve functional upper airway collapsibility during sleep.88-90 Indeed, depending on the level of advancement, on average, MAS therapy improves upper airway collapsibility by 2 to 6 cmH2O.88,90 However, the precise mechanisms mediating these improvements and the reasons why the magnitude of change differs markedly between individuals remains unclear. Nonetheless, the predominant role of MAS therapy is to improve anatomical impairment of the upper airway.MAS and Nonanatomical Phenotypes
In addition to a direct role on anatomical impairment, MAS therapy may also improve upper airway muscle function to reduce OSA severity. There are two key possible mechanisms: (1) MAS therapy may stimulate local reflex afferents to increase upper airway muscle activity or, (2) changes in airway anatomy with MAS therapy may result in increased mechanical efficiency of the airway dilator muscles such that a given level of neural drive results in greater improvements in airflow (improved muscle efficiency). To date, the few studies that have been conducted to investigate these two potential mechanisms have yielded contrasting results. Consistent with the first mechanism, two earlier studies that used surface recording electrodes showed acute increases in upper airway dilator muscle activity (genioglossus and geniohyoid) awake (seated and upright)91 and asleep during respiratory events with mandibular advancement.92 Another study also showed that the baseline activity of the submandibular and masseter muscles measured supine during quiet breathing and the activity of the submandibular and posterior temporalis muscles with mandibular advancement were higher in responders versus nonresponders to MAS therapy.93 However, two more recent studies using gold standard intramuscular electromyography recordings of the genioglossus muscle during sleep did not detect increases in muscle activity with MAS therapy. Rather, one study found that consistent with improved upper airway anatomy, MAS therapy reduced negative epiglottic pressure and consequently genioglossus muscle activity.94 The other study specifically investigated genioglossus muscle responsiveness to negative airway pressure swings and did not detect systematic changes in muscle responsiveness with increasing levels of mandibular advancement.90 However, the sample size was relatively small.Two studies have investigated the second potential mechanism, increased mechanical efficiency with MAS therapy. Edwards and colleagues89 assessed changes in ventilation (upper airway airflow) as a surrogate measure of muscle function. No difference in this parameter was found with the MAS device in place versus without. Bamagoos and colleagues90 systematically investigated changes in minute ventilation and airflow in conjunction with genioglossus EMG in response to experimentally induced airflow limitation using transient reductions in CPAP at different levels of mandibular advancement. Similar to the study by Edwards and colleagues,89 there was no evidence for improved muscle efficacy with mandibular advancement.90 A limitation of both of these studies was that the sample size was quite small; therefore, the potential for some individuals with certain phenotypes to have improved muscle function with MAS therapy remains. Accordingly, additional appropriately designed studies to investigate the effects of MAS therapy on upper airway muscle function are required.
Thus, although there are clear theoretical reasons as to why MAS therapy could improve upper airway muscle function to complement the anatomical benefits, the available evidence to date shows contrasting effects of MAS therapy on upper airway muscle activity and no evidence for systematic improvements in upper airway muscle function. Thus far, there have been limited studies into how MAS therapy alters the other two nonanatomical phenotypes – loop gain and arousal threshold. Edwards and colleagues89 found that wearing a MAS device did not affect these two phenotypes. However, the study participants were already on MAS therapy for varying amounts of time, potentially influencing the expression of these two non-anatomical phenotypes. Thus, future studies in treatment-naïve individuals with OSA would be insightful.
A PHENOTYPIC APPROACH TO OSA THERAPY: THE PALM SCALE
As highlighted, it is evident that OSA is a multifactorial disorder. Nonanatomical phenotypes interact with an underlying anatomical impairment to cause OSA and mediate its severity, or protect against OSA if favorable. The combination of these phenotypes vary between individuals with OSA, with some primarily having an anatomical impairment whereas others may also have impairment in multiple nonanatomical phenotypes.32 These recent insights into the pathophysiology of OSA have led to the development of a scale to characterize the four key phenotypic causes of OSA to inform tailored treatment: the Pcrit, Arousal threshold, Loop gain and Muscle responsiveness (PALM scale).32,33 Briefly, because impaired anatomy is the main driver of OSA, patients are first categorized into one of three groups according to their upper airway anatomy/collapsibility (Pcrit). PALM scale 1 patients (23%) have severely impaired anatomy (Pcrit> + 2 cmH2O) and likely require a major anatomical intervention to treat their OSA (e.g. CPAP). Most individuals with OSA (58%) are categorized as PALM scale 2. These patients have moderately impaired anatomy (Pcrit between -2 and + 2 cmH2O) and are potential candidates for one or a combination of targeted anatomical (such as MAS, upper airway surgery, or supine avoidance device) and/or nonanatomical interventions (e.g., a hypnotic increase the arousal threshold and reduce OSA severity in those with a low arousal threshold; O2 to reduce unstable ventilatory control in those with high loop gain; or a drug to increase pharyngeal muscle contractility in those with poor muscle responsiveness during sleep). Finally, the remaining 19% of patients are PALM scale 3. These individuals have only mild anatomical impairment (Pcrit between -2 and -5cmH2O), similar to many individuals without OSA. All patients categorized as PALM scale 3 have impairment in one or more nonanatomical phenotypes. These patients are candidates for one or more targeted therapies with nonanatomic interventions likely to be particularly beneficial (see Figures 1 and 3). Importantly, using this conceptual framework and the known effect sizes for non-CPAP interventions, it is estimated that more than 50% of all patients with OSA could be treated with one or more non-CPAP interventions if appropriately directed at the abnormal trait(s).95Figure 3Schematic summary of existing and evolving targeted therapies to treat OSA. |
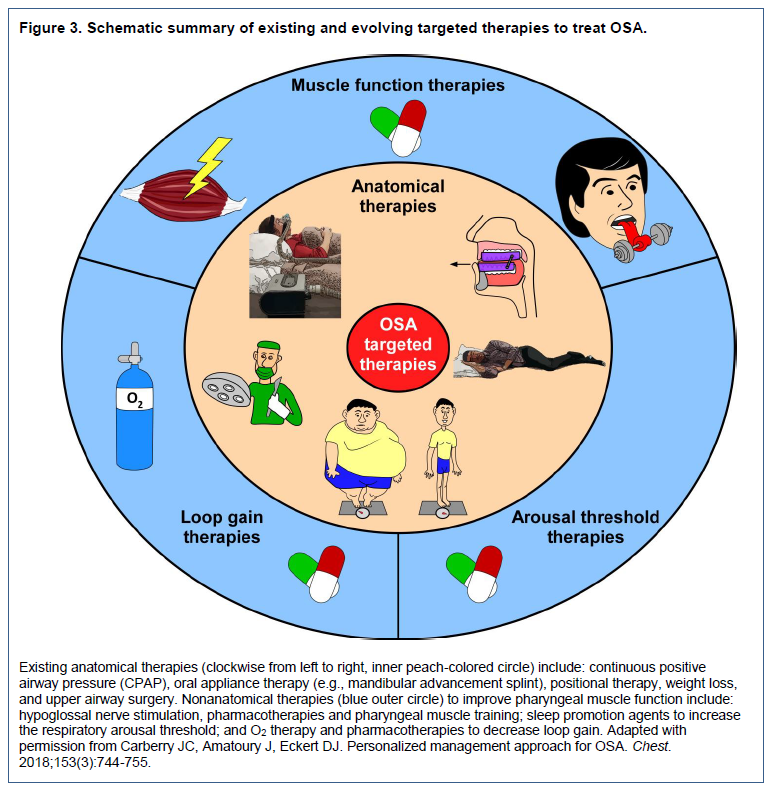{kind=link}
Application of the PALM Scale Approach to MAS Therapy
Consistent with the variable treatment responses to MAS therapy in OSA, the PALM scale predicts that MAS therapy will improve the impaired anatomy phenotype to yield therapeutic benefit in some but not all patients with OSA. Specifically, given that on average MAS therapy improves Pcrit by 2 to 6 cmH2O,88,90 patients who have less impairment in upper airway anatomy at baseline (PALM scale 2 and 3) are expected to have a larger therapeutic response with MAS therapy than those with highly impaired anatomy (patients categorized as PALM scale 1) in whom minimal or no change is expected. Indeed, in order to resolve the anatomical impediment of OSA, the goal of therapy is to reduce Pcrit below -5cmH2O.33 Furthermore, the PALM scale predicts that nonanatomical traits will be important mediators of treatment response to MAS therapy. Specifically, with its primary role to improve upper airway anatomy, and to a lesser extent, a potential role on upper airway muscle function, consistent with the available data,89 MAS therapy is not anticipated to alter the other two nonanatomical traits (loop gain and arosual threshold). Thus, in patients who have high levels of impairment in one or more of the nonanatomical traits (e.g., low arousal threshold or high loop gain), repetitive awakenings and unstable respiratory control, and therefore OSA, are expected to persist with MAS therapy. In support of this mechanistic rationale, a recent retrospective study in which the four phenotypic traits were estimated in 14 patients with OSA with and without MAS therapy demonstrated greater therapeutic efficacy in those with minimal anatomical impairment (mild upper airway collapsibility) and minimal respiratory instability (low loop gain).89 As discussed in the next paragraphs, the current challenge is how to translate these concepts to the clinical setting.TRANSLATION OF OSA PHENOTYPING CONCEPTS TO THE CLINICAL SETTING
To overcome the barriers associated with current gold standard phenotyping techniques (e.g., invasive overnight in-laboratory studies limited to research investigations),33 there have been recent advances in the development of simplified phenotyping tools for clinical implementation. For example, brief breathing tests acquired during wakefulness correlate well with upper airway collapsibility (Pcrit) during sleep.96,97 Similarly, automated measures of peak flow obtained via a standard overnight PSG provide good estimates of Pcrit.98 An individual’s therapeutic CPAP level (≤8.0 cmH2O) also shows good sensitivity (89%) and specificity (84%) to identify a patient with a mildly collapsible upper airway.99 Thus, these approaches may be helpful clinically to differentiate the extent of anatomical impairment (i.e., those with highly versus minimally collapsible airways), the main contributor to OSA, to facilitate targeted treatment decisions using PALM scale concepts. Similarly, inspiratory airflow profiles during flow limitation from an overnight sleep study may provide information on the site of upper airway collapse.100Recent strategies have also been developed to estimate the nonanatomical phenotypes. This includes estimates of loop gain and arousal threshold from PSG recordings using sophisticated analysis methods,101,102 or via simple breath hold maneuvers,103 or simply using standard clinical PSG variables.104 Although a simple clinically useful estimate of upper airway muscle function is yet to be determined, recent advanced signal processing techniques have been used successfully to estimate all four phenotypes including muscle compensation from standard PSG recordings.105
From a clinical perspective, some of these methods rely entirely on data (i.e. AHI, nadir oxygen saturation, fraction of events that are hypopneas, therapeutic CPAP levels) that are already routinely collected during diagnostic or titration sleep studies for OSA.99,104 Thus, these strategies can be incorporated into the current recommended diagnostic pathway for OSA106 with negligible additional cost to help guide patient selection decisions for MAS therapy. Conversely, others still require advanced signal processing analysis or specialized equipment before they can be implemented into the current diagnostic pathway. However, the computations for some of these methods can be completed within 10 minutes for each patient on a standard personal computer and there is scope for automation.101,102 Other methods can be performed relatively quickly during wakefulness and do not require entire overnight PSG.96,97 Therefore, considering the potential health and safety benefits from achieving treatment success via tailored therapy with fewer treatment failures and associated costs (Figure 1), the cost-effectiveness is likely to be superior compared to the current diagnostic pathway. However, formal cost-benefit analyses will clearly be required as phenotyping approaches continue to be developed, refined, and improved. Nonetheless, these concepts and evolving tools offer promise that the goal of clinically feasible, accurate tools to tailor therapy to individual patients with OSA will be a reality in the not-too-distant future. This includes new approaches to optimize MAS therapy and improved prediction of treatment success.
SUMMARY AND CONCLUSIONS
OSA phenotyping concepts offer potential to facilitate realignment of the current treatment management approach for OSA, which is too frequently time consuming, costly, and ineffective. In the context of dental sleep medicine, detailed OSA phenotyping can help to determine the mechanisms of MAS therapy, which may help to optimize MAS therapy design for greater efficacy. Phenotyping approaches also have the potential to assist in the accurate identification of the characteristics of responders to MAS therapy. Indeed, the ideal candidate based on current phenotyping concepts and the available evidence is a patient with a mild to moderately collapsible upper airway with minimal or no impairment in the other non-anatomical phenotypes (i.e., the patient does not have high loop gain). Clinical implementation of these concepts could improve current prediction approaches and reduce MAS therapy failure rates, which has long been a major clinical challenge for the field. Finally, as simplified phenotyping approaches and novel non-CPAP therapies continue to develop and evolve, there is also scope for phenotyping approaches to be used to inform targeted combination therapy for those with major impairment in the nonanatomical phenotypes and in those who do not respond to MAS therapy alone.107REFERENCES
2. Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol. 2013;177(9):1006-1014.
3. Young T, Skatrud J, Peppard PE. Risk factors for obstructive sleep apnea in adults. JAMA. 2004;291(16):2013-2016.
4. Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep M. Clinical Guideline for the Evaluation, Management and Long-term Care of Obstructive Sleep Apnea in Adults. J Clin Sleep Med. 2009;5(3):263-276.
5. Lacasse Y, Godbout C, Sériès F. Health-related quality of life in obstructive sleep apnoea. Eur Resp J. 2002;19(3):499-503.
6. Ellen RL, Marshall SC, Palayew M, Molnar FJ, Wilson KG, Man-Son-Hing M. Systematic review of motor vehicle crash risk in persons with sleep apnea. J Clin Sleep Med. 2006;2(02):193-200.
7. Hla KM, Young T, Hagen EW, et al. Coronary Heart Disease Incidence in Sleep Disordered Breathing: The Wisconsin Sleep Cohort Study. Sleep. 2015;38(5):677-684.
8. Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea–hypopnea and incident stroke: The Sleep Heart Health Study. Am J Resp Crit Care Med. 2010;182(2):269-277.
9. Aurora RN, Punjabi NM. Obstructive sleep apnoea and type 2 diabetes mellitus: a bidirectional association. Lancet Resp Med. 2013;1(4):329-338.
10. Osorio RS, Gumb T, Pirraglia E, et al. Sleep-disordered breathing advances cognitive decline in the elderly. Neurology. 2015;84(19):1964-1971.
11. BaHammam AS, Kendzerska T, Gupta R, et al. Comorbid depression in obstructive sleep apnea: an under-recognized association. Sleep Breath. 2016;20(2):447-456.
12. Marshall NS, Wong KK, Phillips CL, Liu PY, Knuiman MW, Grunstein RR. Is sleep apnea an independent risk factor for prevalent and incident diabetes in the Busselton Health Study? J Clin Sleep Med. 2009;5(1):15-20.
13. McDaid C, Durée KH, Griffin SC, et al. A systematic review of continuous positive airway pressure for obstructive sleep apnoea–hypopnoea syndrome. Sleep Med Rev. 2009;13(6):427-436.
14. Galic T, Bozic J, Pecotic R, Ivkovic N, Valic M, Dogas Z. Improvement of cognitive and psychomotor performance in patients with mild to moderate obstructive sleep apnea treated with mandibular advancement device: A prospective 1-year study. J Clin Sleep Med. 2016;12(2):177-186.
15. Lin HC, Friedman M, Chang HW, Gurpinar B. The efficacy of multilevel surgery of the upper airway in adults with obstructive sleep apnea/hypopnea syndrome. Laryngoscope. 2008;118(5):902-908.
16. Peppard PE, Young T, Palta M, Dempsey J, Skatrud J. Longitudinal study of moderate weight change and sleep-disordered breathing. JAMA. 2000;284(23):3015-3021.
17. McEvoy RD, Antic NA, Heeley E, et al. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med. 2016;375(10):919-931.
18. Weaver TE, Grunstein RR. Adherence to continuous positive airway pressure therapy: the challenge to effective treatment. Proc Am Thor Soc. 2008;5(2):173-178.
19. Sutherland K, Vanderveken OM, Tsuda H, et al. Oral appliance treatment for obstructive sleep apnea: An update. J Clin Sleep Med. 2014;10(2):215-227.
20. Sutherland K, Takaya H, Qian J, Petocz P, Ng AT, Cistulli PA. Oral appliance treatment response and polysomnographic phenotypes of obstructive sleep apnea. J Clin Sleep Med. 2015;11(8):861-868.
21. Lee RW, Petocz P, Prvan T, Chan AS, Grunstein RR, Cistulli PA. Prediction of obstructive sleep apnea with craniofacial photographic analysis. Sleep. 2009;32(1):46-52.
22. Jayawardhana M, Sutherland K, Cistulli P, Chazal P. Prediction of MAS therapy response in obstructive sleep apnoea patients using clinical data. Conf Proc IEEE Eng Med Biol Soc. 2018;2018:6040-6043.
23. Vroegop AV, Vanderveken OM, Dieltjens M, et al. Sleep endoscopy with simulation bite for prediction of oral appliance treatment outcome. J Sleep Res. 2013;22(3):348-355.
24. Phillips CL, Grunstein RR, Darendeliler MA, et al. Health outcomes of continuous positive airway pressure versus oral appliance treatment for obstructive sleep apnea: a randomized controlled trial. Am J Respir Crit Care Med. 2013;187(8):879-887.
25. Carberry JC, Amatoury J, Eckert DJ. Personalized management approach for OSA. Chest. 2018;153(3):744-755.
26. Macey PM, Woo MA, Kumar R, Cross RL, Harper RM. Relationship between obstructive sleep apnea severity and sleep, depression and anxiety symptoms in newly-diagnosed patients. PLoS One. 2010;5(4):e10211.
27. Weaver EM, Woodson BT, Steward DL. Polysomnography indexes are discordant with quality of life, symptoms, and reaction times in sleep apnea patients. Otolaryngol Head Neck Surg. 2005;132(2):255-262.
28. Johansson P, Alehagen U, Svanborg E, Dahlstrom U, Brostrom A. Sleep disordered breathing in an elderly community-living population: Relationship to cardiac function, insomnia symptoms and daytime sleepiness. Sleep Med. 2009;10(9):1005-1011.
29. Muraja-Murro A, Nurkkala J, Tiihonen P, et al. Total duration of apnea and hypopnea events and average desaturation show significant variation in patients with a similar apnea-hypopnea index. J Med Eng Technol. 2012;36(8):393-398.
30. Wu H, Zhan X, Zhao M, Wei Y. Mean apnea-hypopnea duration (but not apnea-hypopnea index) is associated with worse hypertension in patients with obstructive sleep apnea. Medicine. 2016;95(48):e5493.
31. Alex R, Manchikatla S, Machiraju K, et al. Effect of apnea duration on apnea induced variations in cerebral blood flow velocity and arterial blood pressure. Conf Proc IEEE Eng Med Biol Soc. 2014;2014:270-273.
32. Eckert DJ, White DP, Jordan AS, Malhotra A, Wellman A. Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Resp Crit Care Med. 2013;188(8):996-1004.
33. Eckert DJ. Phenotypic approaches to obstructive sleep apnoea – new pathways for targeted therapy. Sleep Med Rev. 2018;37:45-59.
34. Neelapu BC, Kharbanda OP, Sardana HK, et al. Craniofacial and upper airway morphology in adult obstructive sleep apnea patients: A systematic review and meta-analysis of cephalometric studies. Sleep Med Rev. 2017;31:79-90.
35. Kirkness JP, Schwartz AR, Schneider H, et al. Contribution of male sex, age, and obesity to mechanical instability of the upper airway during sleep. J Appl Physiol. 2008;104(6):1618-1624.
36. Dempsey JA, Veasey SC, Morgan BJ, O'Donnell CP. Pathophysiology of sleep apnea. Physiol Rev. 2010;90(1):47-112.
37. Ravesloot MJL, de Vries N. One hundred consecutive patients undergoing drug-induced sleep endoscopy: Results and evaluation. Laryngoscope. 2011;121(12):2710-2716.
38. Vroegop AV, Vanderveken OM, Boudewyns AN, et al. Druginduced sleep endoscopy in sleepdisordered breathing: Report on 1,249 cases. Laryngoscope. 2014;124(3):797-802.
39. Marques M, Genta PR, Azarbarzin A, et al. Retropalatal and retroglossal airway compliance in patients with obstructive sleep apnea. Respir Physiol Neurobiol. 2018;258:98-103.
40. Schwab RJ, Pasirstein M, Pierson R, et al. Identification of upper airway anatomic risk factors for obstructive sleep apnea with volumetric magnetic resonance imaging. Am J Resp Crit Care Med. 2003;168(5):522-530.
41. Gray EL, McKenzie DK, Eckert DJ. Obstructive sleep apnea without obesity is common and difficult to treat: Evidence for a distinct pathophysiological phenotype. J Clin Sleep Med. 2017;13(1):81–88.
42. Okubo M, Suzuki M, Horiuchi A, et al. Morphologic analyses of mandible and upper airway soft tissue by MRI of patients with obstructive sleep apnea hypopnea syndrome. Sleep. 2006;29(7):909-915.
43. Lee RW, Vasudavan S, Hui DS, et al. Differences in craniofacial structures and obesity in Caucasian and Chinese patients with obstructive sleep apnea. Sleep. 2010;33(8):1075-1080.
44. Chi L, Comyn F-L, Mitra N, et al. Identification of craniofacial risk factors for obstructive sleep apnoea using three-dimensional MRI. Eur Resp J. 2011;38(2):348-358.
45. Saboisky JP, Butler JE, Fogel RB, et al. Tonic and phasic respiratory drives to human genioglossus motoneurons during breathing. J Neurophysiol. 2006;95(4):2213-2221.
46. Carberry JC, Hensen H, Fisher LP, et al. Mechanisms contributing to the response of upper-airway muscles to changes in airway pressure. J Appl Physiol. 2015;118(10):1221-1228.
47. Stanchina ML, Malhotra A, Fogel RB, et al. Genioglossus muscle responsiveness to chemical and mechanical stimuli during non-rapid eye movement sleep. Am J Respir Crit Care Med. 2002;165(7):945-949.
48. Eckert DJ, McEvoy RD, George KE, Thomson KJ, Catcheside PG. Genioglossus reflex inhibition to upper-airway negative-pressure stimuli during wakefulness and sleep in healthy males. J Physiol. 2007;581(3):1193-1205.
49. Wilkinson V, Malhotra A, Nicholas CL, et al. Discharge patterns of human genioglossus motor units during sleep onset. Sleep. 2008;31(4):525-533.
50. Carberry JC, Jordan AS, White DP, Wellman A, Eckert DJ. Upper airway collapsibility (Pcrit) and pharyngeal dilator muscle activity are sleep stage dependent. Sleep. 2016;39(3):511-521.
51. Loewen AHS, Ostrowski M, Laprairie J, Maturino F, Hanly PJ, Younes M. Response of genioglossus muscle to increasing chemical drive in sleeping obstructive apnea patients. Sleep. 2011;34(8):1061-1073.
52. Sands SA, Eckert DJ, Jordan AS, et al. Enhanced upper-airway muscle responsiveness is a distinct feature of overweight/obese individuals without sleep apnea. Am J Respir Crit Care Med. 2014;190(8):930-937.
53. Brown EC, Cheng S, McKenzie DK, Butler JE, Gandevia SC, Bilston LE. Respiratory movement of upper airway tissue in obstructive sleep apnea. Sleep. 2013;36(7):1069-1076.
54. Bilston LE, Gandevia SC. Biomechanical properties of the human upper airway and their effect on its behavior during breathing and in obstructive sleep apnea. J Appl Physiol. 2014;116(3):314-324.
55. Saboisky JP, Butler JE, Gandevia SC, Eckert DJ. Functional role of neural injury in obstructive sleep apnea. Front Neurol. 2012;3:95-95.
56. Eckert DJ, Lo YL, Saboisky JP, Jordan AS, White DP, Malhotra A. Sensorimotor function of the upper-airway muscles and respiratory sensory processing in untreated obstructive sleep apnea. J Appl Physiol. 2011;111(6):1644-1653.
57. Camacho M, Certal V, Abdullatif J, et al. Myofunctional therapy to treat obstructive sleep apnea: A systematic review and meta-analysis. Sleep. 2015;38(5):669-675.
58. Strollo PJ, Soose RJ, Maurer JT, et al. Upper-airway stimulation for obstructive sleep apnea. N Engl J Med. 2014;370(2):139-149.
59. Eastwood PR, Barnes M, Walsh JH, et al. Treating obstructive sleep apnea with hypoglossal nerve stimulation. Sleep. 2011;34(11):1479-1486.
60. Horner RL, Grace KP, Wellman A. A resource of potential drug targets and strategic decision-making for obstructive sleep apnoea pharmacotherapy. Respirology. 2017;22(5):861-873.
61. Taranto-Montemurro L, Sands SA, Edwards BA, et al. Desipramine improves upper airway collapsibility and reduces OSA severity in patients with minimal muscle compensation. Eur Resp J. 2016;48(5):1340-1350.
62. Taranto-Montemurro L, Edwards BA, Sands SA, et al. Desipramine Increases Genioglossus Activity and Reduces Upper Airway Collapsibility during Non-REM Sleep in Healthy Subjects. Am J Resp Crit Care Med. 2016;194(7):878-885.
63. Lim R, Carberry JC, Wellman A, Grunstein R, Eckert DJ. Reboxetine and hyoscine butylbromide improve upper airway function during non-REM and suppress REM sleep in healthy individuals. Sleep.
64. Taranto-Montemurro L, Messineo L, Sands SA, et al. The combination of atomoxetine and oxybutynin greatly reduces obstructive sleep apnea severity: A randomized, placebo-controlled, double-blind crossover trial. Am J Resp Crit Care Med. 2018; doi: 10.1164/rccm.201808-1493OC. [Epub ahead of print].
65. Phillipson EA, Sullivan CE. Arousal: The forgotten response to respiratory stimuli. Am Rev Resp Dis. 1978;118(5):807-809.
66. Younes M. Role of Arousals in the pathogenesis of obstructive sleep apnea. Am J Resp Crit Care Med. 2004;169(5):623-633.
67. Eckert DJ, Younes MK. Arousal from sleep: implications for obstructive sleep apnea pathogenesis and treatment. J Appl Physiol. 2014;116(3):302-313.
68. Younes M, Ostrowski M, Atkar R, Laprairie J, Siemens A, Hanly P. Mechanisms of breathing instability in patients with obstructive sleep apnea. J Appl Physiol. 2007;103(6):1929-1941.
69. Eckert DJ, Malhotra A, Wellman A, White DP. Trazodone increases the respiratory arousal threshold in patients with obstructive sleep apnea and a low arousal threshold. Sleep. 2014;37(4):811-819.
70. Eckert DJ, Owens RL, Kehlmann G B, et al. Eszopiclone increases the respiratory arousal threshold and lowers the apnoea/hypopnoea index in obstructive sleep apnoea patients with a low arousal threshold. Clin Sci (Lond). 2011;120(12):505-514.
71. Carter SG, Berger MS, Carberry JC, et al. Zopiclone Increases the arousal threshold without impairing genioglossus activity in obstructive sleep apnea. Sleep. 2016;39(4):757-766.
72. Carberry JC, Fisher LP, Grunstein RR, et al. Role of common hypnotics on the phenotypic causes of obstructive sleep apnoea: paradoxical effects of zolpidem. Eur Respir J. 2017;50(6).
73. Carberry JC, Grunstein R, Eckert DJ. The effects of zolpidem in obstructive sleep apnoea– an open-label pilot study. J Sleep Res. 2019:In press.
74. Berry RB, Kouchi K, Bower J, Prosise G, Light RW. Triazolam in patients with obstructive sleep apnea. Am J Resp Crit Care Med. 1995;151(2):450-454.
75. Carter SG, Carberry JC, Cho G, et al. Effect of 1 month of zopiclone on obstructive sleep apnoea severity and symptoms: a randomised controlled trial. Eur Respir J. 2018;52(1).
76. Edwards BA, Sands SA, Owens RL, et al. The combination of supplemental oxygen and a hypnotic markedly improves obstructive sleep apnea in patients with a mild to moderate upper airway collapsibility. Sleep. 2016;39(11):1973-1983.
77. Osman AM, Carter SG, Carberry JC, Eckert DJ. Obstructive sleep apnea: current perspectives. Nat Sci Sleep. 2018;10:21-34.
78. Younes M, Ostrowski M, Thompson W, Leslie C, Shewchuk W. Chemical control stability in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2001;163(5):1181-1190.
79. Wellman A, Jordan AS, Malhotra A, et al. Ventilatory control and airway anatomy in obstructive sleep apnea. Am J Respir Crit Care Med. 2004;170(11):1225-1232.
80. Wellman A, Malhotra A, Jordan AS, Stevenson KE, Gautam S, White DP. Effect of oxygen in obstructive sleep apnea: role of loop gain. Respir Physiol Neurobiol. 2008;162(2):144-151.
81. Edwards BA, Sands SA, Eckert DJ, et al. Acetazolamide improves loop gain but not the other physiological traits causing obstructive sleep apnoea. J Physiol. 2012;590(5):1199-1211.
82. Chan AS, Sutherland K, Schwab RJ, et al. The effect of mandibular advancement on upper airway structure in obstructive sleep apnoea. Thorax. 2010;65(8):726-732.
83. Tsuiki S, Lowe AA, Almeida FR, Kawahata N, Fleetham JA. Effects of mandibular advancement on airway curvature and obstructive sleep apnoea severity. Eur Resp J. 2004;23(2):263-268.
84. Sam K, Lam B, Ooi CG, Cooke M, Ip MS. Effect of a non-adjustable oral appliance on upper airway morphology in obstructive sleep apnoea. Resp Med. 2006;100(5):897-902.
85. Kyung SH, Park Y-C, Pae E-K. Obstructive sleep apnea patients with the oral appliance experience pharyngeal size and shape changes in three dimensions. Angle Orthod. 2005;75(1):15-22.
86. Brown EC, Cheng S, McKenzie DK, Butler JE, Gandevia SC, Bilston LE. Tongue and lateral upper airway movement with mandibular advancement. Sleep. 2014;36(3):397-404.
87. Cistulli PA, Gotsopoulos H, Marklund M, Lowe AA. Treatment of snoring and obstructive sleep apnea with mandibular repositioning appliances. Sleep Med Rev. 2004;8(6):443-457.
88. Ng AT, Gotsopoulos H, Qian J, Cistulli PA. Effect of oral appliance therapy on upper airway collapsibility in obstructive sleep apnea. Am J Resp Crit Care Med. 2003;168(2):238-241.
89. Edwards BA, Andara C, Landry S, et al. Upper-airway collapsibility and loop gain predict the response to oral appliance therapy in patients with obstructive sleep apnea. Am J Resp Crit Care Med. 2016;194(11):1413-1422.
90. Bamagoos AA, Cistulli PA, Sutherland K, et al. Dose-dependent effects of mandibular advancement on upper airway collapsibility and muscle function in obstructive sleep apnea. Sleep. 2019; Feb 22. pii: zsz049. doi: 10.1093/sleep/zsz049. [Epub ahead of print].
91. Johal A, Gill G, Ferman A, McLaughlin K. The effect of mandibular advancement appliances on awake upper airway and masticatory muscle activity in patients with obstructive sleep apnoea. Clin Physiol Funct Imaging. 2007;27(1):47-53.
92. Yoshida K. Effect of a prosthetic appliance for treatment of sleep apnea syndrome on masticatory and tongue muscle activity. J Prosthet Dent. 1998;79(5):537-544.
93. Ma SY, Whittle T, Descallar J, et al. Association between resting jaw muscle electromyographic activity and mandibular advancement splint outcome in patients with obstructive sleep apnea. Am J Orthod Dentofacial Orthop. 2013;144(3):357-367.
94. Burke P, Knapman F, Tong B, et al. Effects of mandibular advancement splints on upper airway physiology in obstructive sleep apnoea. J Sleep Res. 2018;27(2):P114 [abstract].
95. Owens RL, Edwards BA, Eckert DJ, et al. An integrative model of physiological traits can be used to predict obstructive sleep apnea and response to non positive airway pressure therapy. Sleep. 2015;38(6):961-970.
96. Hirata RP, Schorr F, Kayamori F, et al. Upper airway collapsibility assessed by negative expiratory pressure while awake is associated with upper airway anatomy. J Clin Sleep Med. 2016;12(10):1339-1346.
97. Osman A, Carberry J, Burke PG, Grunstein R, Eckert D. Upper airway collapsibility measured using a simple wakefulness test closely relates to the pharyngeal critical closing pressure (Pcrit) during sleep in obstructive sleep apnea. Sleep.
98. Azarbarzin A, Sands SA, Taranto-Montemurro L, et al. Estimation of pharyngeal collapsibility during sleep by peak inspiratory airflow. Sleep. 2017;40(1).
99. Landry SA, Edwards BA, Hamilton GS, et al. Therapeutic CPAP level predicts upper airway collapsibility in patients with obstructive sleep apnea. Sleep. 2017;40(6).
100. Genta PR, Sands SA, Butler JP, et al. Airflow shape is associated with the pharyngeal structure causing OSA. Chest. 2017;152(3):537-546.
101. Terrill PI, Edwards BA, Nemati S, et al. Quantifying the ventilatory control contribution to sleep apnoea using polysomnography. Eur Respir J. 2015;45(2):408-418.
102. Sands SA, Terrill PI, Edwards BA, et al. Quantifying the arousal threshold using polysomnography in obstructive sleep apnea. Sleep. 2018;41(1).
103. Messineo L, Taranto-Montemurro L, Azarbarzin A, et al. Breath-holding as a means to estimate the loop gain contribution to obstructive sleep apnoea. J Physiol. 2018;596(17):4043-4056.
104. Edwards BA, Eckert DJ, McSharry DG, et al. Clinical Predictors of the Respiratory Arousal Threshold in Patients with Obstructive Sleep Apnea. Am J Resp Crit Care Med. 2014;190(11):1293-1300.
105. Sands SA, Edwards BA, Terrill PI, et al. Phenotyping pharyngeal pathophysiology using polysomnography in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2018;197(9):1187-1197.
106. Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: An American Academy of Sleep Med clinical practice guideline. J Clin Sleep Med. 2017;13(3):479-504.
107. Lai V, Tong B, Tran C, et al. Combination therapy with mandibular advancement and expiratory positive airway pressure valves reduces OSA severity. J Sleep Res. 2018;27(2):P168 [abstract].
SUBMISSION & CORRESPONDENCE INFORMATION
Submitted for publication March 4, 2019
Submitted in final revised form March 28, 2019
Accepted for publication March 29, 2019
Address correspondence to: Prof. Danny J. Eckert, Adelaide Institute for Sleep Health (AISH), Flinders University, Level 2, Mark Oliphant Building, 5 Laffer Drive, Bedford Park SA 5042, Phone: +61 8 7421 9780, Email: danny.eckert@flinders.edu.au
DISCLOSURE STATEMENT
DJE is supported by a National Health and Medical Research Council of Australia Senior Research Fellowship (1116942), has a Cooperative Research Centre (CRC)-P grant: a collaborative grant between the Australian Government, Academia and Industry (Industry partner Oventus Medical), has been a collaborator on research projects in which SomnoMed and Zephyr have provided equipment, has research grants from Apnimed and Bayer and serves on the Scientific Advisory Board for Apnimed. VL and JCC have no potential conflicts to declare.
PDF